Doença é causada por um enfraquecimento e destruição das células vermelhas do sangue. A talassemia surge por genes ou variantes que afetam a forma como o corpo produz a hemoglobina, proteína presente nos glóbulos vermelhos e que transporta oxigênio.
Talassemia pode causar complicações significativas, incluindo pneumonia, sobrecarga de ferro, deformidades ósseas e doença cardiovascular. No entanto, a condição hereditária dos glóbulos vermelhos confere um grau de proteção contra a malária, que é ou foi predominante nas regiões onde o traço da doença é considerado comum.
Epidemiologia da Talassemia
A forma de beta talassemia está prevalente entre os povos do Mediterrâneo. Essa associação geográfica é responsável pela nomeação. Na Europa, as maiores concentrações da doença são encontradas na Grécia, nas regiões costeiras da Turquia (particularmente a região do mar Egeu, como Izmir, Balikesir, Aydin, Mugla e Mediterrâneo, como Antalya, Adana e Mersin) e no Sul da Itália.
As principais ilhas do Mediterrâneo (exceto as Baleares) como a Sicília, Sardenha, Malta, Córsega, Chipre e Creta são afetadas em particular. Outros povos da região, bem como aqueles nas proximidades, também apresentam altas taxas de talassemia, incluindo residentes do oeste da Ásia e norte da África.
Talassemia
Longe do Mediterrâneo, sul-asiáticos também são afetados, com segunda maior concentração mundial de portadores (16% da população). Hoje em dia é encontrada em populações que vivem na África, nas Américas e Nepal.
Talassemias são associadas com pessoas de origem mediterrânea, os árabes (em especial os palestinos e os povos de descendência palestina) e asiáticos. As Maldivas tem a maior incidência no mundo, com uma taxa de portador de 18% da população.
A prevalência estimada é de 16% em pessoas a partir do Chipre, 1% na Tailândia, e 3-8% na população de Bangladesh, China, Índia, Malásia e Paquistão. Talassemias também ocorrem em descendentes de pessoas da América Latina e dos países mediterrânicos (por exemplo, Grécia, Itália, Portugal, Espanha, entre outros).
Fisiopatologia da Talassemia
Normalmente, a hemoglobina é composta por quatro cadeias proteicas, duas globinas α e duas β. Em talassemia, os pacientes têm defeitos em qualquer cadeia de globina de acordo com grande parte dos especialistas.
Beta-Talassemia
Beta-talassemias ocorrem devido às mutações no gene HBB ao cromossomo 11, também herdado de forma autossômica recessiva. A gravidade da doença depende da natureza da mutação.
Complicações da Talassemia
Sobrecarga de ferro: Pessoas com talassemia têm uma sobrecarga de ferro em seus corpos, da própria doença ou de transfusões de sangue. O excesso resulta em danos para o sistema de coração, fígado e endócrino, que inclui as glândulas produtoras de hormonas reguladoras dos processos do corpo.
O dano é caracterizado por depósitos excessivos de ferro. Sem tratamento adequado, quase todos os pacientes com beta-talassemia acumulam os níveis de FE fatais em potencial.
Infecções: Pessoas com talassemia têm um risco aumentado de infecção. Isto é especialmente verdadeiro se o baço foi removido.
Deformidades Ósseas: Talassemia pode fazer a medula óssea se expandir e os ossos alargarem. Isto pode resulta em estrutura óssea anormal, em especial na face e no crânio. Expansão torna a estrutura fina e quebradiça, aumentando o risco de fraturas em níveis consideráveis.
Saiba Mais
Baço: Auxilia no combate às infecções e filtros de materiais indesejados, tais como células sanguíneas, velhas ou danificadas. A talassemia é por vezes acompanhada pela destruição de um grande número de células vermelhas do sangue e a tarefa de remover as mesmas pode fazer com que o baço aumente. Esplenomegalia deixa a anemia piorar e reduzir a vida útil da qualidade do sangue transfundido. Alargamento grave exige a remoção.
Anemia pode causar crescimento de uma criança a abrandar. A puberdade também é adiada em pessoas com talassemia.
Problemas Cardíacos: Insuficiência cardíaca congestiva e ritmos cardíacos anormais (arritmias) estão associados com a talassemia grave.
Assistência Médica
Talassemia Leve: Pacientes com traços não necessitam de cuidados médicos ou follow-up após o diagnóstico inicial ser feito. Apenas advertidos que a condição é diagnosticada pela anemia por deficiência de ferro.
Aconselhamento é indicado em todas as pessoas com doenças genéticas, especialmente quando a família está em risco de uma forma grave que pode ser prevenida.
Talassemia Grave: Os pacientes com talassemia grave necessitam de tratamento médico. Um regime de transfusão de sangue foi à primeira medida eficaz no prolongamento da vida.






Tratamentos Medicamentosos
Os pacientes com talassemia acumulam de modo gradual altos níveis de ferro (Fe) em seus corpos. Esta acumulação pode ser devido à própria doença ou por várias transfusões de sangue recebidas. Essa sobrecarga traz consigo certas complicações bioquímicas.
Dois principais agentes envolvidos no transporte de ferro e armazenamento no corpo são ferritina e transferrina. A ferritina é uma proteína presente nas células, que se liga ao Fe (II) e armazena na forma de Fe (III), libertando no sangue sempre que necessário.
Transferrina é uma proteína de ligação ao ferro presente no plasma sanguíneo. Atua como transportador para o fornecimento de células com o metal através de endocitose. Ela tem características específicas para FE (III), conforme aponta grande parte dos profissionais especialistas.






O Que É Talassemia?
A talassemia é uma doença do sangue transmitida através de famílias (herdado), na qual o corpo produz forma anormal de hemoglobina, a proteína dos glóbulos vermelhos que transporta oxigênio. O distúrbio resulta em destruição excessiva das células vermelhas do sangue e leva para a condição de anemia.
A hemoglobina é composta de duas proteínas: Alfa globina e beta globina. Talassemia ocorre quando existe um defeito no gene que ajuda a produção de uma destas proteínas de controle.
Existem Dois Tipos Principais de Talassemia
- A talassemia alfa ocorre quando um gene ou genes relacionados com a proteína de globina alfa estão ausentes ou alteradas.
- Talassemia beta acontece nos momentos em que os defeitos genéticos semelhantes afetam a produção da proteína.
- Talassemias beta ocorre em pessoas de origem mediterrânea, em menor medida entre chineses, asiáticos e afro-americanos.
- A forma mais grave de alfa talassemia causa natimorto (morte do feto durante o nascimento ou os estágios finais da gravidez).
Outros Sintomas Podem Incluir:
- Deformidades ósseas na face
- Fadiga
- Falta de crescimento
- Falta de ar
- Pele amarelada (icterícia)












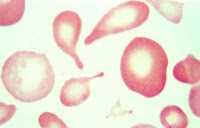
Estudo microscopia de campo escuro, contraste de fase e campo claro. Tenho interesse sobre o assunto relacionado acima.
MUito bom!
Obrigada!
Paz e luz!
Vânia Maria